treats manual
2024-08-28
1 treats: trees and traits simulations.
Simultaneously simulate phylogenetic trees and traits (and the interaction between them).
The core of this package is based on the diversitree birth-death algorithm.
1.1 What is treats?
Bib is studying the evolution of mammalian disparity (i.e. diversity of shapes) across the Cretaceous-Palaeogene mass extinction event (K-Pg - 66 million years ago). They observe a decrease of morphological disparity after the extinction event followed by a sharp increase of disparity. However Bib is also prudent and wants to test whether their observed pattern is just an artifact of the data or actually showing some effect of the K-Pg extinction on mammalian evolution. First they need to check whether changes in disparity can actually be discernables due to an extinction event (i.e. if an extinction event happens, would it be possible to detect a change in disparity)? Second they want to know whether these changes are due to the mass extinction event being selective or not (i.e. the mass extinction favored some traits).
The illustrative example section actually shows how do implement this example in treats in details.
Of course, this is just an example of when to use treats.
Simulating trees and traits can also be useful for:
- Provide a baseline/null model to compare to an observed one.
- Testing evolutionary hypotheses.
- Exploring statistics properties (i.e. can the disparity metric X capture changes in Y).
- Easily generating illustrative/simplified evolutionary history examples.
- Teaching simplified macro-evolutionary theory (e.g. the birth-death model).
Note that for approaching these points individually (or some aspects of it), there are some very good R packages that simulate birth-death trees (e.g. TreeSim) or simulate traits (disparity) and diversity jointly (e.g. RPANDA or PETER). We strongly advise you look at these packages first as they might be more appropriate for your needs.
treats is a highly modular and user-friendly package that contains the simulations functionality of all these packages: it allows you to simulate disparity and diversity jointly with a vast array of options that can be easily modified by users.
For example, you can easily generate any type of process to generate a trait (BM, OU, something else, etc.) in multiple dependent or independent dimensions through "traits" objects.
You can then specify how the traits should affect disparity through "modifiers" objects.
And finally you can create events (like mass extinctions) through "events" objects.
These objects and how to modify them will be detailed throughout this manual.
Finally we are putting an emphasis in the development of this package on the speed and reliability of the functions.
1.1.1 Modular?
Because there is an infinite number of ways you might want to generate disparity and diversity (different traits, different modifiers and different events), the treats package is designed to make all these parts easy to code separately and integrate in the treats core functions.
This allows you to simulate finely-tuned multidimensional data for your specific project!
1.2 Who is this manual for?
This manual explains how treats works in as much details as possible and aims to give you the keys to design your own diversity and disparity simulations.
The first section Getting started is aimed for people with a R beginner level:
- you know what a package is - and how to install it;
- you know what a function is - and what arguments are;
- you have already (vaguely) heard that there are different types of objects in R - like matrices (
"matrix") or trees ("phylo"); - and you have already looked for documentation online.
The sections after that are a bit more advanced and require the following level:
- you have already created functions before;
- you know how to subset elements of a list - i.e. you understand the
list$elementsyntax; - you are aware that both
TRUE == 1andFALSE == 0areTRUE; - you have heard that the objects in R have classes and that it sometimes matters - for example you know that R can differentiate between
as.numeric(1)andas.integer(1); - you owe part of your work to Stack Overflow.
1.3 Installing and running the package
You can install this package easily, directly from the GitHub:
## Checking if devtools is already installed
if(!require(devtools)) install.packages("devtools")
## Installing the latest version directly from GitHub
devtools::install_github("TGuillerme/treats")1.4 Help
If you need help with the package, hopefully the following manual will be useful. However, if you have suggestions or comments on on what has already been developed or will be developed, please send me an email (guillert@tcd.ie) or if you are a GitHub user, directly create an issue on the GitHub page. Doing so will not only help you but also other users as it will help improve this manual and the package!
1.5 How does treats work?
Basically, the treats function takes your traits, modifiers and events objects to simulate disparity and diversity.
You will find more details about how these objects (traits, modifiers and events) work in the rest of the tutorial but here is a graphical representation of how treats works:
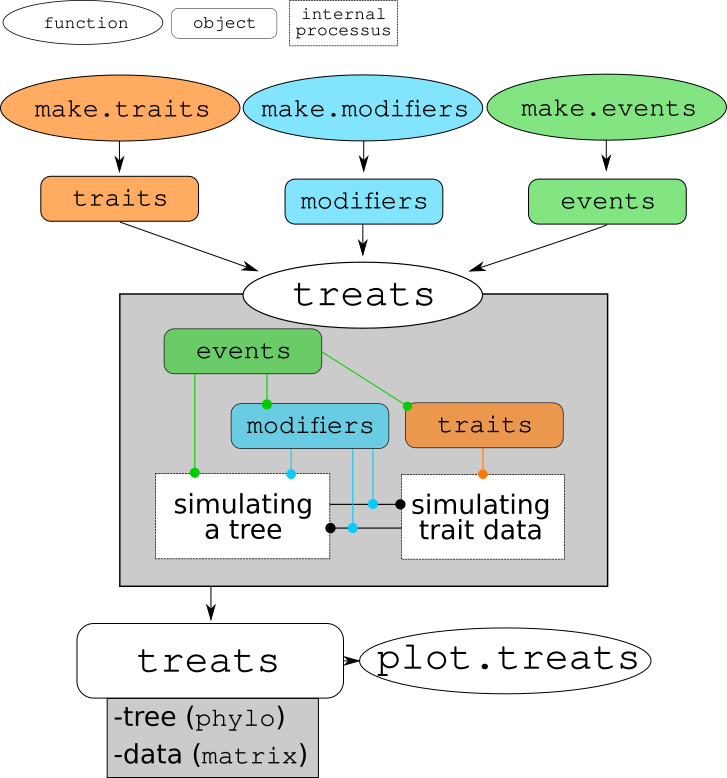
Schematic summary of the treats package architecture
1.6 The birth-death algorithm
If you want to get your hands dirty, you can go straight to the getting started section, this following section just describes the process in pseudo-code.
The treats algorithm is based on a modular version of the birth-death process algorithm.
The birth-death model is a continuous Markov process (i.e. a continuous random process where future events depend only on the present, and not on past events) which is well defined mathematically and commonly used in evolutionary biology but also in many other fields.
The algorithm used in treats allows modularity of this process and is based on the following steps (the text in courier font is for the name of the process in the algorithm):
- Starting the process: this step is non-modular and creates a random tree with one tip, one node and one branch connecting both. This step is used to optimise the rest of the algorithm in terms of speed and memory management. The node, tip and branch resulting from this step are discarded at the end of the simulations.
- Selecting a lineage (
selecting): this step selects a tip that is currently not extinct. In a standard birth-death process this is done randomly, however intreatsthis can be modified based on the birth-death parameters, the currently available lineages and potential trait values. For example, it is possible to put a higher probability for selecting a lineage that is closely related to a lineage that recently went extinct and has a positive trait value. Then go to step 2. - Growing the tree (
waiting): this step grows the tree by a certain amount. It does so by adding the same amount of branch length to all the non-extinct lineages. In an exact birth-death process, this is done by drawing a random value from an exponential distribution with a rate of \[\text{number of living lineages} \times (\text{speciation} + \text{extinction parameters})\]. Intreats, this can be modified based on the birth-death parameters, the currently available lineages and potential trait values. For example, it is possible to increase branch length by the ratio of living/extinct fossils and a random number drawn from the range of current trait values. If the total tree length is less than the required tree length, go to step 3. Else go to step 6. - Simulating traits [optional](
traits): this step allows you to simulate a trait value for the selected lineage from step 1. This is typically not part of a standard birth-death process and is handled via thetraitsoption intreats(see themake.traitschapter for more details). Then go to step 4. - Speciating (
speciating): in this step, the selected lineage from step 1 has the option of speciating or going extinct. In a standard birth-death process, this happens by randomly drawing a value between 0 and 1 as a way to trigger speciation relative to the birth-death parameters:
if
randomly drawn number is smaller or equal to speciation/(speciation + extinction)
then
do speciate
else
go extinctAgain, in treats, this process can be modified based on the birth-death parameters, the currently available lineages and potential trait values. For example, the lineage can only speciate if its trait value is positive, regardless of whether speciation or extinction have been triggered.
Then go to step 5.
5. If the number of lineages is less than required number of lineages, then go to step 1. Else go to step 6.
6. The simulation stops because it has reached the required amount of lineages and/or time (i.e. branch length).
## Loading required package: dispRity